8.5. Global Mechanisms
Global mechanisms provide a framework to have certain reactions work on specific subsets of species in a CRN, applied at the end of compilation to all species created by Component and Mixture Mechanisms.
Under the hood, global mechanisms work just like normal mechanisms - they take a set of species and compile them into CRNs. Global mechanisms are called at the end of the compilation, so they apply to all species generated by all local mechanisms.
Keywords:
filter_dict {str : True/False} is used to make global mechanisms selective. The dictionaries keys (strings) can be species’ names, types, or attributes. The mechanism is then applied or not based upon the value of that attribute in the filter_dict. For exampe filter_dict = {“dna”:False} would not apply its mechanism to any “dna” species.
default_on True / False: For species with no attributes in the filter_dict, a global mechanism defaults to its default_on keyword, which can be True or False.
recursive_species_filtering True / False: When applying a filter dictionary to a ComplexSpecies, this keyword determines if the filter will be applied recursively to all species inside that complex species. For example, consider the filter dict filter_dict = {“dna”:False}. A ComplexSpecies (material_type=”complex”) consisting of a species of type material_type=”dna” and a species of type material_type=”protein” will not be effected by this filter unless recursive_species_filtering = True. Note that attributes are automatically inheritted recursively, so this keyword only matters for name and material_type filters.
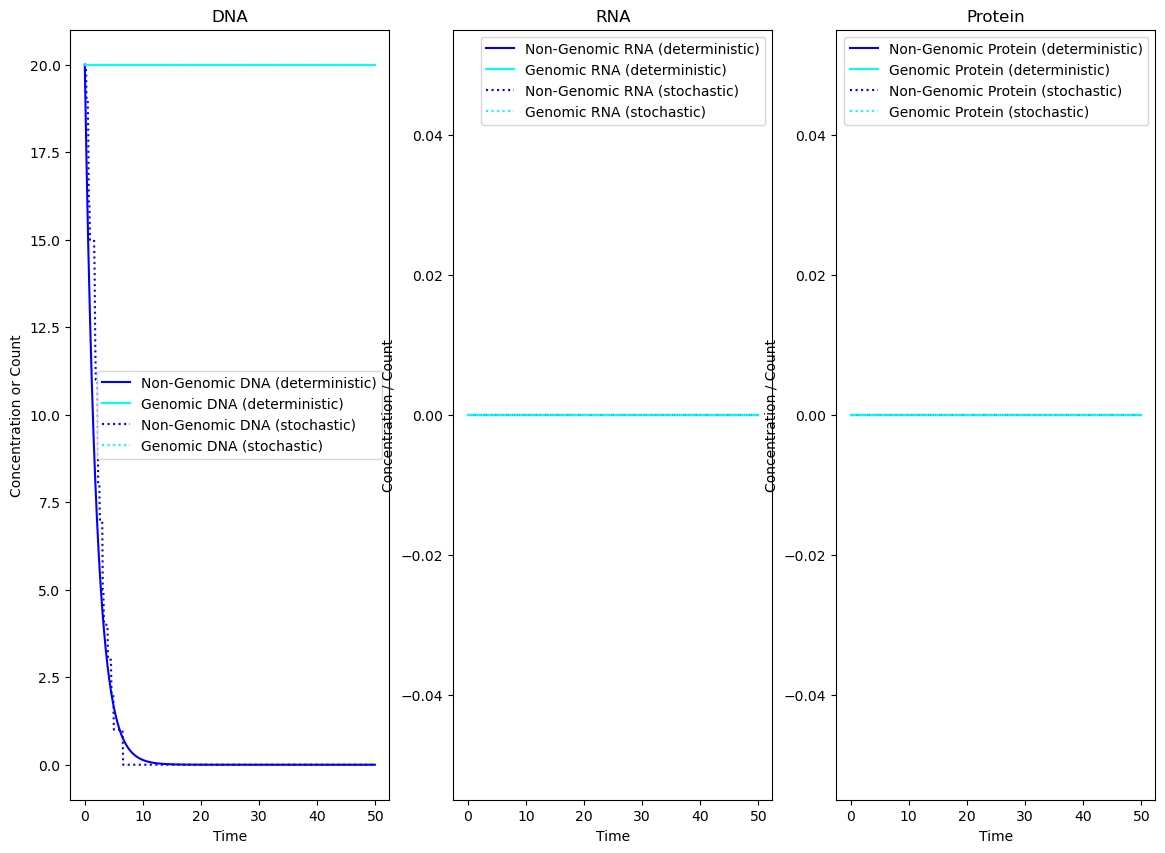
Example 1: Global Dilution using attributes
In the following example, a model will be set up where global mechanisms cause degradation by dilution on all species which are do not have the attributes “genomic” or “machinery”
[1]:
from biocrnpyler.components import DNAassembly
from biocrnpyler.mechanisms import Dilution
from biocrnpyler.mixtures import TxTlExtract
#We will only use default parameters in this model, for simplicity.
kb, ku, ktx, ktl, kdeg, kdil = 100, 20, 3, 2, .5, .5
parameters = {"kb":kb, "ku":ku, "ktx":ktx, "ktl":ktl, "kdeg": kdeg, "kdil":kdil}
#Creates a global dilution mechanism that acts on all species generated except for
# those with the type or attribute "genome" or "machinery"
dilution_mechanism = Dilution(filter_dict = {"genomic":False, "machinery":False}, default_on = True)
#Add this mechanism to a dictionary which is passed into the Mixture txtl.TxTlExtract
global_mechanisms = {"dilution":dilution_mechanism}
myMixture = TxTlExtract(name = "txtl", parameters = parameters, global_mechanisms = global_mechanisms)
#Add machinery attributes to species I want constiutively expressed at the dilution rate
myMixture.rnap.add_attribute("machinery")
myMixture.rnaase.add_attribute("machinery")
myMixture.ribosome.add_attribute("machinery")
#Creates a dna assembly. This assembly is type "dna" so it will be degraded
A_dna = DNAassembly(name = "G1", promoter = "pBest", rbs = "BCD2")
#Create another dna assembly but set its internal specie's attributes to contain "genomic" so it will not be degraded
#Note: this only protects the dna_G2 species encoded by this assembly as well as complex species (eg rnap:DNA) which inherit their subspecies attributes.
A_genome = DNAassembly(name = "G2", promoter = "pBest", rbs = "BCD2", attributes = ["genomic"])
myMixture.add_components(A_dna)
myMixture.add_components(A_genome)
myCRN = myMixture.compile_crn()
print(myCRN.pretty_print(show_rates=True, show_material=True,
show_attributes=True, show_keys=False))
print("Simulating with BioSCRAPE")
try:
print("Simulating with BioSCRAPE")
import numpy as np
import pylab as plt
timepoints = np.arange(0, 50, .1)
x0_dict = {myMixture.ribosome.get_species():100,
myMixture.rnap.get_species():20,
myMixture.rnaase.get_species():10,
A_dna.dna:20,
A_genome.dna:20}
full_result_sto = myCRN.simulate_with_bioscrape_via_sbml(timepoints,
initial_condition_dict = x0_dict,
stochastic = True)
full_result_det = myCRN.simulate_with_bioscrape_via_sbml(timepoints,
initial_condition_dict = x0_dict,
stochastic = False)
if (full_result_det is not None) and (full_result_sto is not None):
#chemical_reaction_network.get_all_species_containing is a useful shortcut to get lists of species
tot_A_dna_det = np.sum(full_result_det[myCRN.get_all_species_containing(A_dna.dna, return_as_strings=True)], 1)
tot_A_genome_det = np.sum(full_result_det[myCRN.get_all_species_containing(A_genome.dna, return_as_strings=True)], 1)
tot_A_dna_sto = np.sum(full_result_sto[myCRN.get_all_species_containing(A_dna.dna, return_as_strings=True)], 1)
tot_A_genome_sto = np.sum(full_result_sto[myCRN.get_all_species_containing(A_genome.dna, return_as_strings=True)], 1)
tot_A_dna_rna_det = np.sum(full_result_det[myCRN.get_all_species_containing(A_dna.transcript, return_as_strings=True)], 1)
tot_A_genome_rna_det = np.sum(full_result_det[myCRN.get_all_species_containing(A_genome.protein, return_as_strings=True)], 1)
tot_A_dna__rna_sto = np.sum(full_result_sto[myCRN.get_all_species_containing(A_dna.transcript, return_as_strings=True)], 1)
tot_A_genome_rna_sto = np.sum(full_result_sto[myCRN.get_all_species_containing(A_genome.protein, return_as_strings=True)], 1)
plt.figure(figsize = (14, 10))
plt.subplot(131)
plt.plot(timepoints, tot_A_dna_det, color = "blue", label = "Non-Genomic DNA (deterministic)")
plt.plot(timepoints, tot_A_genome_det, color = "cyan", label = "Genomic DNA (deterministic)")
plt.plot(timepoints, tot_A_dna_sto, ":", color = "blue", label = "Non-Genomic DNA (stochastic)")
plt.plot(timepoints, tot_A_genome_sto, ":", color = "cyan", label = "Genomic DNA (stochastic)")
plt.legend()
plt.xlabel("Time")
plt.title("DNA")
plt.ylabel("Concentration or Count")
plt.subplot(132)
plt.plot(timepoints, tot_A_dna_rna_det, color = "blue", label = "Non-Genomic RNA (deterministic)")
plt.plot(timepoints, tot_A_genome_rna_det, color = "cyan", label = "Genomic RNA (deterministic)")
plt.plot(timepoints, tot_A_dna__rna_sto, ":", color = "blue", label = "Non-Genomic RNA (stochastic)")
plt.plot(timepoints, tot_A_genome_rna_sto, ":", color = "cyan", label = "Genomic RNA (stochastic)")
plt.legend()
plt.xlabel("Time")
plt.title("RNA")
plt.ylabel("Concentration / Count")
plt.subplot(133)
plt.plot(timepoints, full_result_det[str(A_dna.protein)], color = "blue", label = "Non-Genomic Protein (deterministic)")
plt.plot(timepoints, full_result_det[str(A_genome.protein)], color = "cyan", label = "Genomic Protein (deterministic)")
plt.plot(timepoints, full_result_sto[str(A_dna.protein)], ":", color = "blue", label = "Non-Genomic Protein (stochastic)")
plt.plot(timepoints, full_result_sto[str(A_genome.protein)], ":", color = "cyan", label = "Genomic Protein (stochastic)")
plt.legend()
plt.title("Protein")
plt.xlabel("Time")
plt.ylabel("Concentration / Count")
plt.show()
except ModuleNotFoundError:
print('please install the plotting libraries: pip install biocrnpyler[all]')
Species(N = 17) = {
complex[protein[Ribo]:rna[G2]] (@ 0),
complex[protein[Ribo]:rna[G1]] (@ 0),
complex[protein[RNAase]:rna[G2]] (@ 0),
complex[protein[RNAase]:rna[G1]] (@ 0),
complex[dna[G2]:protein[RNAP]] (@ 0),
complex[dna[G1]:protein[RNAP]] (@ 0),
complex[complex[protein[Ribo]:rna[G2]]:protein[RNAase]] (@ 0),
complex[complex[protein[Ribo]:rna[G1]]:protein[RNAase]] (@ 0),
protein[Ribo] (@ 0),
protein[RNAase] (@ 0),
protein[RNAP] (@ 0),
protein[G2] (@ 0),
rna[G2] (@ 0),
dna[G2(genomic)] (@ 0),
protein[G1] (@ 0),
rna[G1] (@ 0),
dna[G1] (@ 0),
}
Reactions (27) = [
0. dna[G1]+protein[RNAP] <--> complex[dna[G1]:protein[RNAP]]
Kf=k_forward * dna_G1 * protein_RNAP
Kr=k_reverse * complex_dna_G1_protein_RNAP_
k_forward=100
k_reverse=20
1. complex[dna[G1]:protein[RNAP]] --> dna[G1]+rna[G1]+protein[RNAP]
Kf=k_forward * complex_dna_G1_protein_RNAP_
k_forward=3
2. rna[G1]+protein[Ribo] <--> complex[protein[Ribo]:rna[G1]]
Kf=k_forward * rna_G1 * protein_Ribo
Kr=k_reverse * complex_protein_Ribo_rna_G1_
k_forward=100
k_reverse=20
3. complex[protein[Ribo]:rna[G1]] --> rna[G1]+protein[G1]+protein[Ribo]
Kf=k_forward * complex_protein_Ribo_rna_G1_
k_forward=2
4. dna[G2(genomic)]+protein[RNAP] <--> complex[dna[G2]:protein[RNAP]]
Kf=k_forward * dna_G2_genomic * protein_RNAP
Kr=k_reverse * complex_dna_G2_genomic_protein_RNAP_
k_forward=100
k_reverse=20
5. complex[dna[G2]:protein[RNAP]] --> dna[G2(genomic)]+rna[G2]+protein[RNAP]
Kf=k_forward * complex_dna_G2_genomic_protein_RNAP_
k_forward=3
6. rna[G2]+protein[Ribo] <--> complex[protein[Ribo]:rna[G2]]
Kf=k_forward * rna_G2 * protein_Ribo
Kr=k_reverse * complex_protein_Ribo_rna_G2_
k_forward=100
k_reverse=20
7. complex[protein[Ribo]:rna[G2]] --> rna[G2]+protein[G2]+protein[Ribo]
Kf=k_forward * complex_protein_Ribo_rna_G2_
k_forward=2
8. complex[protein[Ribo]:rna[G2]] -->
Kf=k_forward * complex_protein_Ribo_rna_G2_
k_forward=0.5
9. protein[G1] -->
Kf=k_forward * protein_G1
k_forward=0.5
10. complex[dna[G1]:protein[RNAP]] -->
Kf=k_forward * complex_dna_G1_protein_RNAP_
k_forward=0.5
11. dna[G1] -->
Kf=k_forward * dna_G1
k_forward=0.5
12. protein[G2] -->
Kf=k_forward * protein_G2
k_forward=0.5
13. complex[protein[Ribo]:rna[G1]] -->
Kf=k_forward * complex_protein_Ribo_rna_G1_
k_forward=0.5
14. rna[G2] -->
Kf=k_forward * rna_G2
k_forward=0.5
15. protein[RNAP] -->
Kf=k_forward * protein_RNAP
k_forward=0.5
16. protein[RNAase] -->
Kf=k_forward * protein_RNAase
k_forward=0.5
17. protein[Ribo] -->
Kf=k_forward * protein_Ribo
k_forward=0.5
18. rna[G1] -->
Kf=k_forward * rna_G1
k_forward=0.5
19. complex[protein[Ribo]:rna[G2]]+protein[RNAase] <--> complex[complex[protein[Ribo]:rna[G2]]:protein[RNAase]]
Kf=k_forward * complex_protein_Ribo_rna_G2_ * protein_RNAase
Kr=k_reverse * complex_complex_protein_Ribo_rna_G2__protein_RNAase_
k_forward=100
k_reverse=20
20. complex[complex[protein[Ribo]:rna[G2]]:protein[RNAase]] --> protein[Ribo]+protein[RNAase]
Kf=k_forward * complex_complex_protein_Ribo_rna_G2__protein_RNAase_
k_forward=0.5
21. complex[protein[Ribo]:rna[G1]]+protein[RNAase] <--> complex[complex[protein[Ribo]:rna[G1]]:protein[RNAase]]
Kf=k_forward * complex_protein_Ribo_rna_G1_ * protein_RNAase
Kr=k_reverse * complex_complex_protein_Ribo_rna_G1__protein_RNAase_
k_forward=100
k_reverse=20
22. complex[complex[protein[Ribo]:rna[G1]]:protein[RNAase]] --> protein[Ribo]+protein[RNAase]
Kf=k_forward * complex_complex_protein_Ribo_rna_G1__protein_RNAase_
k_forward=0.5
23. rna[G2]+protein[RNAase] <--> complex[protein[RNAase]:rna[G2]]
Kf=k_forward * rna_G2 * protein_RNAase
Kr=k_reverse * complex_protein_RNAase_rna_G2_
k_forward=100
k_reverse=20
24. complex[protein[RNAase]:rna[G2]] --> protein[RNAase]
Kf=k_forward * complex_protein_RNAase_rna_G2_
k_forward=0.5
25. rna[G1]+protein[RNAase] <--> complex[protein[RNAase]:rna[G1]]
Kf=k_forward * rna_G1 * protein_RNAase
Kr=k_reverse * complex_protein_RNAase_rna_G1_
k_forward=100
k_reverse=20
26. complex[protein[RNAase]:rna[G1]] --> protein[RNAase]
Kf=k_forward * complex_protein_RNAase_rna_G1_
k_forward=0.5
]
Simulating with BioSCRAPE
Simulating with BioSCRAPE
/Users/murray/Library/CloudStorage/Dropbox/macosx/src/biocrnpyler/biocrnpyler/core/chemical_reaction_network.py:512: UserWarning: Trying to set species that is not in model: protein_Ribo_machinery
m.set_species(processed)
/Users/murray/Library/CloudStorage/Dropbox/macosx/src/biocrnpyler/biocrnpyler/core/chemical_reaction_network.py:512: UserWarning: Trying to set species that is not in model: protein_RNAP_machinery
m.set_species(processed)
/Users/murray/Library/CloudStorage/Dropbox/macosx/src/biocrnpyler/biocrnpyler/core/chemical_reaction_network.py:512: UserWarning: Trying to set species that is not in model: protein_RNAase_machinery
m.set_species(processed)
/Users/murray/Library/CloudStorage/Dropbox/macosx/src/biocrnpyler/biocrnpyler/core/chemical_reaction_network.py:512: UserWarning: Trying to set species that is not in model: protein_Ribo_machinery
m.set_species(processed)
/Users/murray/Library/CloudStorage/Dropbox/macosx/src/biocrnpyler/biocrnpyler/core/chemical_reaction_network.py:512: UserWarning: Trying to set species that is not in model: protein_RNAP_machinery
m.set_species(processed)
/Users/murray/Library/CloudStorage/Dropbox/macosx/src/biocrnpyler/biocrnpyler/core/chemical_reaction_network.py:512: UserWarning: Trying to set species that is not in model: protein_RNAase_machinery
m.set_species(processed)
Example 2: Global Dilution using material_type with and without recursive_species_filtering
In this example a very simple model of a piece of DNA \(G\) binding to a protein \(P\) to form a complex will be considered. Dilution will be applied to all species excent those of type “dna”. When recursive_species_filtering is False, the protein AND the DNA-protein complex are both diluted. When recursive_species_filtering is True, only the protein is diluted.
[2]:
from biocrnpyler.core import Species
from biocrnpyler.components import ChemicalComplex
from biocrnpyler.mixtures import ExpressionExtract
G = Species("G", material_type = "dna")
A = Species("A", material_type = "protein")
C1 = ChemicalComplex([G, A])
dilution_mechanism_no_recursion = Dilution(filter_dict = {"dna":False}, default_on = True, recursive_species_filtering = False)
M_no_recursion = ExpressionExtract(components = [C1], global_mechanisms = {"dilution":dilution_mechanism_no_recursion}, parameters = parameters)
CRN_no_recursion = M_no_recursion.compile_crn()
print("CRN: recursive_species_filtering = False:\n", CRN_no_recursion)
dilution_mechanism_recursion = Dilution(filter_dict = {"dna":False}, default_on = True, recursive_species_filtering = True)
M_recursion = ExpressionExtract(components = [C1], global_mechanisms = {"dilution":dilution_mechanism_recursion}, parameters = parameters)
CRN_recursion = M_recursion.compile_crn()
print("\nCRN: recursive_species_filtering = True:\n", CRN_recursion.pretty_print(show_keys = True))
CRN: recursive_species_filtering = False:
Species = protein_A, dna_G, complex_dna_G_protein_A_
Reactions = [
protein[A]+dna[G] <--> complex[dna[G]:protein[A]]
protein[A] -->
complex[dna[G]:protein[A]] -->
]
CRN: recursive_species_filtering = True:
Species(N = 3) = {
complex[dna[G]:protein[A]] (@ 0),
dna[G] (@ 0),
protein[A] (@ 0),
}
Reactions (2) = [
0. protein[A]+dna[G] <--> complex[dna[G]:protein[A]]
Kf=k_forward * protein_A * dna_G
Kr=k_reverse * complex_dna_G_protein_A_
k_forward=100
found_key=(mech=None, partid=None, name=kb).
search_key=(mech=one_step_binding, partid=dna_G_protein_A, name=kb).
k_reverse=20
found_key=(mech=None, partid=None, name=ku).
search_key=(mech=one_step_binding, partid=dna_G_protein_A, name=ku).
1. protein[A] -->
Kf=k_forward * protein_A
k_forward=0.5
found_key=(mech=None, partid=None, name=kdil).
search_key=(mech=global_degradation_via_dilution, partid=protein_A, name=kdil).
]
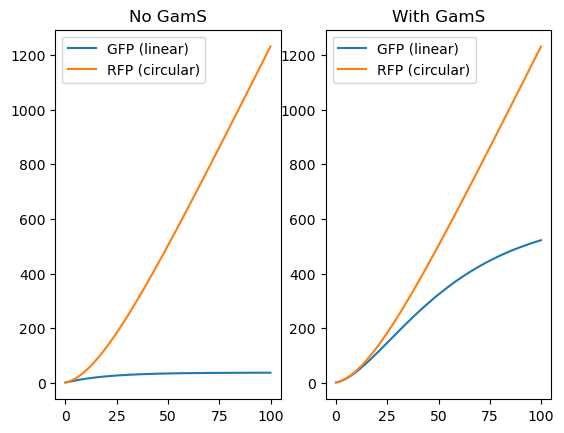
Example 3: Global Degradation of Linear DNA Strands by RecBCD inhibited by GamS
In this example, the degradation of linear DNA strands in an extract is modeled along with the possibility of inhibiting this degradation with GamS, as described by Sun et al. 2014.
This model will use a global degradation mechanism that applies to all DNA without the attribute ‘circular’. This example will create a warning because some Species will have both material_type = ‘dna’ and ‘circular’ in which case the default_on = False keyword will be used to turn off degradation.
[3]:
from biocrnpyler.mechanisms import Deg_Tagged_Degradation
from biocrnpyler.mixtures import SimpleTxTlExtract
parameters = {"kb": 100, "ku": 10,"kdeg": .5, "ktx": .5, "ktl": 1.5, "kdil": .05}
recBCD = Species("recBCD")
gamS = Species("GamS")
linear_dna_degradation = Deg_Tagged_Degradation(
degradase=recBCD, filter_dict={"dna":True, "circular":False}, default_on=False)
inhibited_recBCD = ChemicalComplex([recBCD]+2*[gamS])
Assembly_linear = DNAassembly("GFP", promoter="strong", rbs="weak", initial_concentration=1.0)
Assembly_circular = DNAassembly("RFP", promoter="strong", rbs="weak", initial_concentration=1.0, attributes=["circular"])
M = SimpleTxTlExtract("Mixture 1",
components = [inhibited_recBCD, Assembly_linear, Assembly_circular],
global_mechanisms = [linear_dna_degradation], parameters = parameters)
CRN = M.compile_crn()
print(CRN.pretty_print())
try:
%matplotlib inline
print("Simulating with BioSCRAPE")
import numpy as np
import pylab as plt
timepoints = np.arange(0, 100, .1)
#Simulate with no gamS
x0 = {str(recBCD):1, str(gamS):0, str(Assembly_linear.dna):1.0, str(Assembly_circular.dna):1.0}
R = CRN.simulate_with_bioscrape_via_sbml(timepoints, initial_condition_dict = x0)
#Simulate with GamS
x0 = {str(recBCD):1, str(gamS):20, str(Assembly_linear.dna):1.0, str(Assembly_circular.dna):1.0}
R2 = CRN.simulate_with_bioscrape_via_sbml(timepoints, initial_condition_dict = x0)
if R is not None and R2 is not None:
plt.subplot(121)
plt.title("No GamS")
plt.plot(timepoints, R[str(Assembly_linear.protein)], label = "GFP (linear)")
plt.plot(timepoints, R[str(Assembly_circular.protein)], label = "RFP (circular)")
plt.legend()
plt.subplot(122)
plt.title("With GamS")
plt.plot(timepoints, R2[str(Assembly_linear.protein)], label = "GFP (linear)")
plt.plot(timepoints, R2[str(Assembly_circular.protein)], label = "RFP (circular)")
plt.legend()
plt.show()
except ModuleNotFoundError:
print('please install the plotting libraries: pip install biocrnpyler[all]')
Species(N = 10) = {
protein[RFP] (@ 1.0),
found_key=(mech=initial concentration, partid=None, name=RFP).
search_key=(mech=initial concentration, partid=Mixture 1, name=RFP).
rna[RFP] (@ 1.0),
found_key=(mech=initial concentration, partid=None, name=RFP).
search_key=(mech=initial concentration, partid=Mixture 1, name=RFP).
dna[RFP(circular)] (@ 1.0),
found_key=(mech=initial concentration, partid=None, name=RFP).
search_key=(mech=initial concentration, partid=Mixture 1, name=RFP).
protein[GFP] (@ 1.0),
found_key=(mech=initial concentration, partid=None, name=GFP).
search_key=(mech=initial concentration, partid=Mixture 1, name=GFP).
rna[GFP] (@ 1.0),
found_key=(mech=initial concentration, partid=None, name=GFP).
search_key=(mech=initial concentration, partid=Mixture 1, name=GFP).
dna[GFP] (@ 1.0),
found_key=(mech=initial concentration, partid=None, name=GFP).
search_key=(mech=initial concentration, partid=Mixture 1, name=GFP).
recBCD (@ 0),
complex[dna[GFP]:recBCD] (@ 0),
complex[2x_GamS:recBCD] (@ 0),
GamS (@ 0),
}
Reactions (9) = [
0. 2GamS+recBCD <--> complex[2x_GamS:recBCD]
Kf=k_forward * GamS^2 * recBCD
Kr=k_reverse * complex_GamS_2x_recBCD_
k_forward=100
found_key=(mech=None, partid=None, name=kb).
search_key=(mech=one_step_binding, partid=GamS_2x_recBCD, name=kb).
k_reverse=10
found_key=(mech=None, partid=None, name=ku).
search_key=(mech=one_step_binding, partid=GamS_2x_recBCD, name=ku).
1. dna[GFP] --> dna[GFP]+rna[GFP]
Kf=k_forward * dna_GFP
k_forward=0.5
found_key=(mech=None, partid=None, name=ktx).
search_key=(mech=simple_transcription, partid=strong, name=ktx).
2. rna[GFP] --> rna[GFP]+protein[GFP]
Kf=k_forward * rna_GFP
k_forward=1.5
found_key=(mech=None, partid=None, name=ktl).
search_key=(mech=simple_translation, partid=weak, name=ktl).
3. dna[RFP(circular)] --> dna[RFP(circular)]+rna[RFP]
Kf=k_forward * dna_RFP_circular
k_forward=0.5
found_key=(mech=None, partid=None, name=ktx).
search_key=(mech=simple_transcription, partid=strong, name=ktx).
4. rna[RFP] --> rna[RFP]+protein[RFP]
Kf=k_forward * rna_RFP
k_forward=1.5
found_key=(mech=None, partid=None, name=ktl).
search_key=(mech=simple_translation, partid=weak, name=ktl).
5. dna[GFP]+recBCD <--> complex[dna[GFP]:recBCD]
Kf=k_forward * dna_GFP * recBCD
Kr=k_reverse * complex_dna_GFP_recBCD_
k_forward=100
found_key=(mech=None, partid=None, name=kb).
search_key=(mech=deg_tagged_degradation, partid=dna_GFP, name=kb).
k_reverse=10
found_key=(mech=None, partid=None, name=ku).
search_key=(mech=deg_tagged_degradation, partid=dna_GFP, name=ku).
6. complex[dna[GFP]:recBCD] --> recBCD
Kf=k_forward * complex_dna_GFP_recBCD_
k_forward=0.5
found_key=(mech=None, partid=None, name=kdeg).
search_key=(mech=deg_tagged_degradation, partid=dna_GFP, name=kdeg).
7. rna[GFP] -->
Kf=k_forward * rna_GFP
k_forward=0.05
found_key=(mech=None, partid=None, name=kdil).
search_key=(mech=rna_degradation, partid=rna_GFP, name=kdil).
8. rna[RFP] -->
Kf=k_forward * rna_RFP
k_forward=0.05
found_key=(mech=None, partid=None, name=kdil).
search_key=(mech=rna_degradation, partid=rna_RFP, name=kdil).
]
Simulating with BioSCRAPE
/Users/murray/Library/CloudStorage/Dropbox/macosx/src/biocrnpyler/biocrnpyler/mechanisms/global_mechanisms.py:106: UserWarning: species dna_RFP_circular has multiple attributes(or material type) which conflict with global mechanism filter deg_tagged_degradation. Using default value False.
warn(
[4]:
# End