1. Introduction
This chapter provides a brief introduction to BioCRNpyler. A more detailed introduction to the package is available in [Poo+22].
1.1. Motivation and Background
Chemical reaction networks (CRNs) are commonly used for modeling in systems and synthetic biology [Alo19], [DVM14]. The power of CRNs lies in their expressivity; CRN models can range from physically realistic descriptions of individual molecules to coarse-grained idealizations of complex multi-step processes [DVM14]. However, this expressivity comes at a cost – frequently choosing the right level of detail in a model is more an art than a science. The modeling process requires careful consideration of the desired use of the model, the available data to parameterize the model, and prioritization of certain aspects of modeling or analysis over others.
The available tools for a CRN modeller are vast and include: extensive software to generate and simulate CRNs, databases of models, model analysis tools, and many more [SB22], [Som+15], [LeN+06], [Hoo+06], [Cho+18]. However, relatively few tools exist to aid in the automated construction of CRN models from simple specifications. For example, even though synthetic biologists have taken a module and part-driven approach to their laboratory work [BS05], models are still typically built by hand on a case-by-case basis.
The BioCRNpyler package is a software framework and library designed to aid in the rapid construction of models from common motifs, such as molecular components, biochemical mechanisms, and parameter sets. These parts can be reused and recombined to rapidly generate CRN models in diverse chemical contexts at varying levels of model complexity. Some similar tools exist including [Mye+09], [Har+16], [Tuz+13].
What makes BioCRNpyler unique is an open-source and object-oriented framework written in Python which allows complete control over model compilation by developers as well as a large library of easy-to-use parts and models relevant to synthetic biologists and bio-engineers. The BioCRNpyler package is available on GitHub [BCP-GH].
1.2. The BioCRNpyler Framework
BioCRNpyler is an open-source Python framework that compiles high-level specifications into detailed CRN models saved as SBML [Huc+23]. Specifications may include: biomolecular Components, modeling assumptions (Mechanisms), biochemical context (Mixtures), and Parameters. BioCRNpyler is written in Python with a flexible object-oriented design, extensive documentation, and detailed examples to allow for easy model construction by modelers as well as customization and extension by developers.
Figure 1.1 shows the primary elements of the framework and how they relate to teach other:
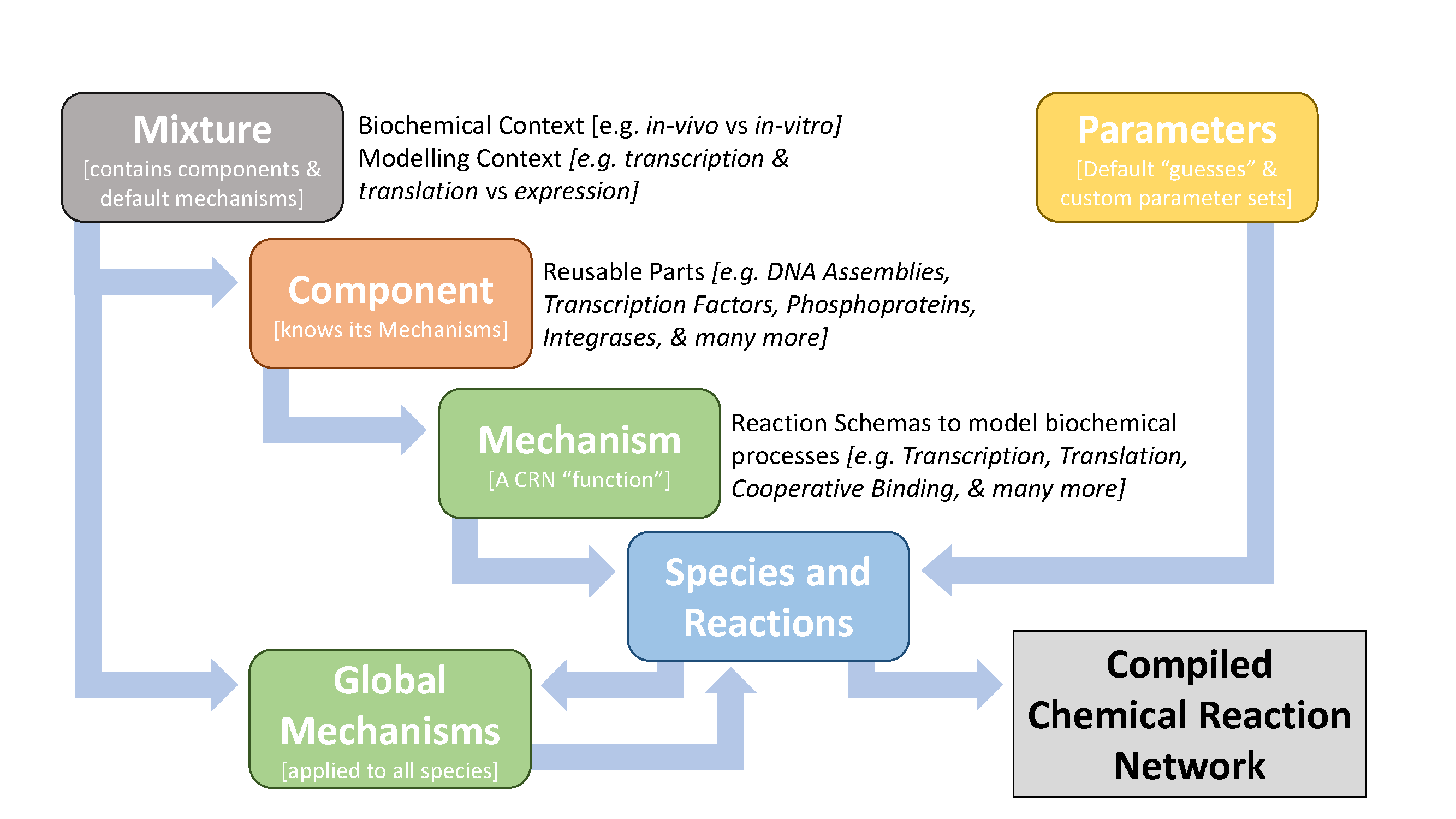
Figure 1.1. The hierarchical organization of classes in the BioCRNpyler framework. Arrows represent compilation.
Species and Reactions make up a CRN and are the output of
BioCRNpyler compilation. Many sub-classes exist such as
ComplexSpecies and reactions with different
kinds of rate function (e.g. mass-action, Hill functions, etc).
Mechanisms are reaction schemas, which can be thought of as abstract functions that produce CRN Species and Reactions. They represent a particular molecular process, such as transcription or translation. During compilation, Mechanisms are called by Components. Global Mechanisms are called at the end of compilation in order to affect all species of a given type or with given attributes — for example, dilution of all protein Species.
Components are reusable parts; they know what kinds of Mechanisms affect them but are agnostic to the underlying schema. For example, a promoter is a Component which will call a transcription Mechanism; similarly, a Ribosome Binding Site (RBS) is a Component which will call a translation Mechanism. However, the same Promoter and RBS can use many different transcription and translation Mechanisms depending on the modeling context and detail desired.
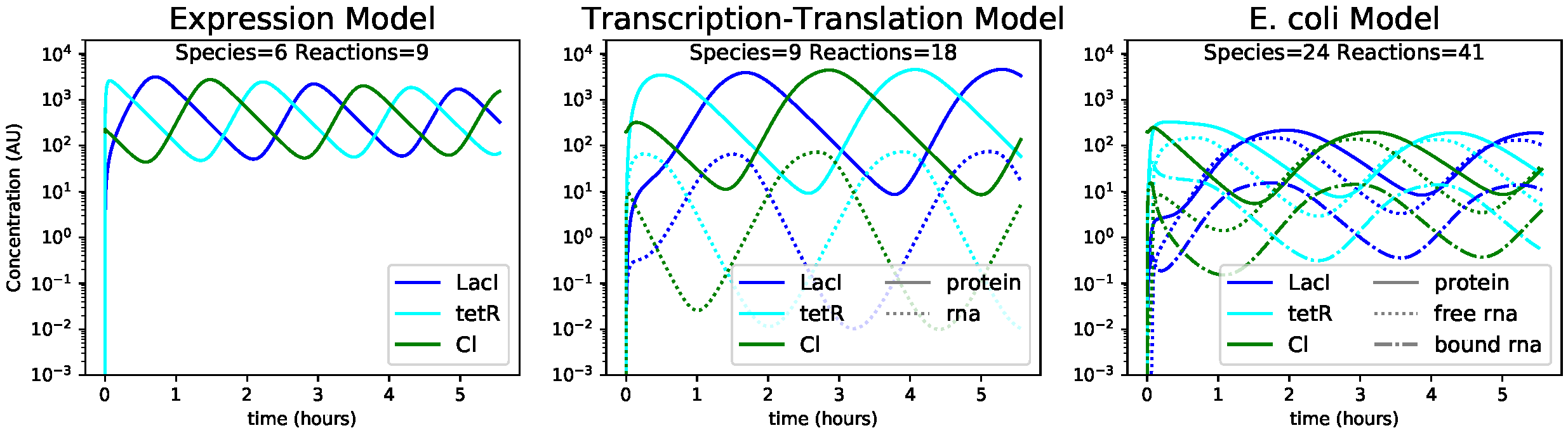
Figure 1.2. Using BioCRNpyler to compile the repressilator at various levels of detail. Simulation parameters come from the repressilator paper [Elo+00] and [Cer+15], [Mil+15]. Simulations were carried out with Bioscrape [Swa+19].
Mixtures are sets of default Mechanisms and Components that represent different molecular and modeling contexts. As an example of molecular context, a cell-extract model requires reactions to consume a finite supply of fuel, while a steady-state model of living cells does not have a limited fuel supply. As an example of modeling context, a simple model of gene expression may have a gene catalytically create a protein product, while a more complex model might include cellular machinery such as RNA polymerase and ribosomes with Michaelis-Menten kinetics.
Parameters are designed for flexibility; they can default to biophysically plausible values (such as a default binding rate), be shared between Components and Mechanisms, or have specific values for Component-Mechanism combinations. This system is designed so that models can be produced quickly without full knowledge of all parameters and then refined with detailed parameter files later.
Compartments are optional in BioCRNpyler. The default compartment
for all species in BioCRNpyler is called “default”. Component Compartments
can be set using setter functions for each component. Membrane components
in BioCRNpyler in biocrnpyler/core/components/membrane/` usually create
an “Internal” and an “External” compartment. To replace the “default”
compartment during compilation of a CRN with compile_crn, a
keyword argument compartment=new_compartment can be passed.
Refer to the examples/Specialized Tutorials/ for example use cases.

Figure 1.3. Python code generating three repressilator CRNs.
1.3. The BioCRNpyler Library
The BioCRNpyler library contains a growing collection of Mechanisms, Components, and Mixtures as well as extensive Jupyter notebooks. Currently, this library is geared towards synthetic biological applications with numerous Mechanisms for transcription, translation, gene regulation, catalysis, molecular binding and many more. Components include common synthetic biological parts such as Promoters, RBSs which can be combined into DNA-assemblies to produce RNA and Proteins, as well as more specific parts such as dCas9.
Mixtures include both models of cell-like systems growing at steady
state and extract-like systems with finite resources. Importantly,
for different modeling contexts, BioCRNpyler includes Mixtures with
different default levels of complexity. The ease in generating
increasingly complex models is illustrated in Figure 1.3, which shows code to compile a repressilator from a few
common Components into multiple CRNs of very different levels of
complexity. Simulations from these models are shown in Figure
1.2.
The latest list of items in the biocrnpyler library can be found on the library page.
1.4. Documentation Conventions
This documentation has a number of notional conventions and functionality:
The left panel displays the table of contents and is divided into two main sections: the User Guide, which contains a narrative description of the package along with examples, and the Reference Manual, which contains documentation for all functions, classes, configurable default parameters, and other detailed information.
Classes, functions, and methods with additional documentation appear in a bold, code font that links to the Reference Manual. Example:
Species.Links to other sections appear in blue. Example: Mechanisms.
Parameters appear in a (non-bold) code font, as do code fragments. Example:
mechanism_type.Example code is contained in code blocks that can be copied using the copy icon in the top right corner of the code block. Code blocks are of three primary types: summary descriptions, code listings, and executed commands.
Summary descriptions show the calling structure of commands but are not directly executable. Example:
rxn = bcp.Reaction(inputs=[s1, s2, ...], outputs=[s3])
Code listings consist of executable code that can be copied and pasted into a Python execution environment. In most cases the objects required by the code block will be present earlier in the file or, occasionally, in a different section or chapter (with a reference near the code block). All code listings assume that the NumPy package is available using the prefix
npand the bioCRNpyler package is imported using prefixbcp. Example:import biocrnpyler as bcp A = bcp.Species('A') B = bcp.Species('B') rxn = bcp.Reaction( inputs=[A], outputs=[B], propensity_type=bcp.MassAction(1e-2) )
Executed commands show commands preceded by a prompt string of the form “>>> “ and also show the output that is obtained when executing that code. The copy functionality for these blocks is configured to only copy the commands and not the prompt string or outputs. Example:
>>> print(rxn.pretty_print()) A --> B Kf=k_forward * A k_forward=0.01
1.5. References
U. Alon, An Introduction to Systems Biology: Design Principles of Biological Circuits. CRC Press, 2019.
Benner SA, Sismour AM. Synthetic biology. Nature Reviews Genetics. 2005;6(7):533–543.
Ceroni F, Algar R, Stan GB, Ellis T, Quantifying cellular capacity identifies gene expression designs with reduced burden. Nature Methods. 2015 May;12(5):415-8. doi: 10.1038/nmeth.3339.
Choi K, Medley JK, König M, Stocking K, Smith L, Gu S, et al. Tellurium: an extensible python- based modeling environment for systems and synthetic biology. Biosystems. 2018;171:74–79.
D. D. Vecchio and R. M. Murray. Biomolecular Feedback Systems. Princton University Press, 2014.
Elowitz MB, et al. A synthetic oscillatory network of transcriptional regulators. Nature. 2000;403(6767):335–338.
Harris LA, et al. BioNetGen 2.2: advances in rule-based modeling. Bioinformatics. 2016;32(21):3366–3368.
S. Hoops, S. Sahle, R. Gauges, C. Lee, J. Pahle, N. Simus, M. Singha l, L. Xu, P. Mendes, U. Kummer, COPASI—a COmplex PAthway SImulator, Bioinformatics. 22(24):3067–3074, 2006. https://doi.org/10.1093/bioinformatics/btl485
Hucka M, et al. The systems biology markup language (SBML): a medium for representation and exchange of biochemical network models. Bioinformatics. 2003;19(4):524–531.
Le Novere N, Bornstein B, Broicher A, Courtot M, Donizelli M, Dharuri H, et al. BioModels Database: a free, centralized database of curated, published, quantitative kinetic models of biochemical and cellular systems. Nucleic acids research. 2006;34(suppl_1):D689–D691.
Milo R, et al. Cell biology by the numbers. Garland Science; 2015.
Myers CJ, et al. iBioSim: a tool for the analysis and design of genetic circuits. Bioinformatics. 2009;25(21):2848–2849.
Poole W, Pandey A, Shur A, Tuza ZA, Murray RM (2022) BioCRNpyler: Compiling chemical reaction networks from biomolecular parts in diverse contexts. PLOS Computational Biology 18(4): e1009987. https://doi.org/10.1371/journal.pcbi.1009987
BioCRNpyler Github Repository; 2025. https://github.com/BuildACell/BioCRNpyler.
The MathWorks, Inc. MATLAB Simbiology Toolbox; 2022. Available from: https://www. mathworks.com/help/simbio/.
Somogyi ET, Bouteiller JM, Glazier JA, König M, Medley JK, Swat MH, et al. libRoadRunner: a high performance SBML simulation and analysis library. Bioinformatics. 2015;31(20):3315–3321.
Swaminathan A, et al. Fast and flexible simulation and parameter estimation for synthetic biology using bioscrape. bioRxiv. 2019; p. 121152.
Tuza ZA, et al. An in silico modeling toolbox for rapid prototyping of circuits in a biomolecular “breadboard” system. In: 52nd IEEE Conference on Decision and Control; 2013. p. 1404–1410.