8.4. Building Gene Regulatory Networks with BioCRNpyler
In this notebook, we will use RegulatedPromoter and CombinatorialPromoter to model Gene Regulation via Transcription Factors.
Note: For this notebook to function, you need the default_parameter.txt file in the same folder as the notebook.
Modeling Gene Regulation by a Single Transcription Factor
In biology, it is very common for a transcription factor to turn on or off promoter. BioCRNpyler has a number of Promoter Components to do this.
RegulatedPromoter: A list of regulators each bind individually to turn a Promoter ON or OFF (No Combinatorial Logic)
RepressiblePromoter: A single repressor modelled with a hill function
ActivatablePromoter: A single activator modeled with a hill function
In the next example, we will produce and compare the outputs of these kinds of promoters.
Example 1: ActivatablePromoter
A very simple Promoter Component modelled with a hill function. However, this class is not able to accurately capture the binding of Machinery like RNAP and shouldn’t be used with Mixtures that include machinery.
[1]:
from biocrnpyler.core import Species
from biocrnpyler.components import DNAassembly, ActivatablePromoter
from biocrnpyler.mixtures import SimpleTxTlExtract
#ActivatedPromoter Example
activator = Species("activator", material_type = "small_molecule")
S_A = Species("A")
#Create a custom set of parameters
hill_parameters = {"k":1.0, "n":4, "K":20, "kleak":.01}
#By Loading custom parameters into the promoter, we override the default parameters of the Mixture
P_activatable = ActivatablePromoter("P_activtable", activator = activator, leak = True, parameters = hill_parameters)
#Create a DNA assembly "reporter" with P_activatable for its promoter
activatable_assembly = DNAassembly(name="activatable_assembly", promoter=P_activatable, rbs="Strong", protein = S_A)
M = SimpleTxTlExtract(name="SimpleTxTl", parameter_file = "default_parameters.txt", components=[activatable_assembly])
CRN = M.compile_crn();
print(CRN.pretty_print(show_rates = True, show_keys = True))
/Users/murray/Library/CloudStorage/Dropbox/macosx/src/biocrnpyler/biocrnpyler/core/parameter.py:678: UserWarning: parameter file contains no unit column! Please add a column named ['unit', 'units'].
warn(
Species(N = 4) = {
small_molecule[activator] (@ 0),
rna[activatable_assembly] (@ 0),
dna[activatable_assembly] (@ 0),
A (@ 0),
}
Reactions (4) = [
0. dna[activatable_assembly] --> dna[activatable_assembly]+rna[activatable_assembly]
Kf = k dna[activatable_assembly] small_molecule[activator]^n / ( K^n + small_molecule[activator]^n )
k=1.0
found_key=(mech=None, partid=None, name=k).
search_key=(mech=positivehill_transcription, partid=P_activtable_activator, name=k).
K=20
found_key=(mech=None, partid=None, name=K).
search_key=(mech=positivehill_transcription, partid=P_activtable_activator, name=K).
n=4
found_key=(mech=None, partid=None, name=n).
search_key=(mech=positivehill_transcription, partid=P_activtable_activator, name=n).
1. dna[activatable_assembly] --> dna[activatable_assembly]+rna[activatable_assembly]
Kf=k_forward * dna_activatable_assembly
k_forward=0.01
found_key=(mech=None, partid=None, name=kleak).
search_key=(mech=positivehill_transcription, partid=P_activtable_activator, name=kleak).
2. rna[activatable_assembly] --> rna[activatable_assembly]+A
Kf=k_forward * rna_activatable_assembly
k_forward=0.25
found_key=(mech=simple_translation, partid=None, name=ktl).
search_key=(mech=simple_translation, partid=Strong, name=ktl).
3. rna[activatable_assembly] -->
Kf=k_forward * rna_activatable_assembly
k_forward=0.001
found_key=(mech=rna_degradation, partid=None, name=kdil).
search_key=(mech=rna_degradation, partid=rna_activatable_assembly, name=kdil).
]
[2]:
#Titrate the activator and plot the result
try:
import bioscrape
import matplotlib.pyplot as plt
except ModuleNotFoundError:
print('please install the plotting libraries: pip install biocrnpyler[all]')
else:
%matplotlib inline
import numpy as np
import pandas as pd
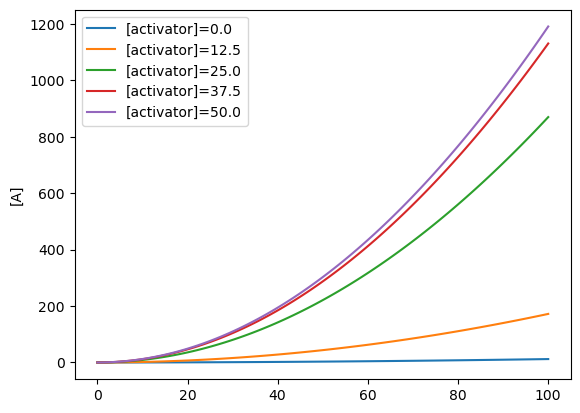
for a_c in np.linspace(0, 50, 5):
x0 = {activatable_assembly.dna:1, activator:a_c}
timepoints = np.linspace(0, 100, 100)
R = CRN.simulate_with_bioscrape_via_sbml(timepoints, initial_condition_dict = x0)
plt.plot(R["time"], R[str(S_A)], label = "[activator]="+str(a_c))
plt.ylabel(f"[{S_A}]")
plt.legend()
plt.show()
Example 2: RepressiblePromoter
A very simple Promoter Component modelled with a hill function. However, this class is not able to accurately capture the binding of Machinery like RNAP and shouldn’t be used with Mixtures that include machinery.
[3]:
from biocrnpyler.components import RepressiblePromoter
#ActivatedPromoter Example
repressor = S_A #defined in the previous example
reporter = Species("reporter", material_type = "protein")
#Create a custom set of parameters
hill_parameters = {"k":1.0, "n":4, "K":20, "kleak":.01}
#By Loading custom parameters into the promoter, we override the default parameters of the Mixture
P_repressible = RepressiblePromoter("P_repressible", repressor = repressor, leak = True, parameters = hill_parameters)
#Create a DNA assembly "reporter" with P_activatable for its promoter
repressible_assembly = DNAassembly(name="reporter", promoter=P_repressible, rbs="Strong", protein = reporter)
M = SimpleTxTlExtract(name="SimpleTxTl", parameter_file = "default_parameters.txt", components=[repressible_assembly])
CRN = M.compile_crn()
print(CRN.pretty_print(show_rates = True, show_keys = True))
Species(N = 4) = {
protein[reporter] (@ 0),
rna[reporter] (@ 0),
dna[reporter] (@ 0),
A (@ 0),
}
Reactions (4) = [
0. dna[reporter] --> dna[reporter]+rna[reporter]
Kf = k dna[reporter] / ( 1 + (A/K)^4 )
k=1.0
found_key=(mech=None, partid=None, name=k).
search_key=(mech=negativehill_transcription, partid=P_repressible_A, name=k).
K=20
found_key=(mech=None, partid=None, name=K).
search_key=(mech=negativehill_transcription, partid=P_repressible_A, name=K).
n=4
found_key=(mech=None, partid=None, name=n).
search_key=(mech=negativehill_transcription, partid=P_repressible_A, name=n).
1. dna[reporter] --> dna[reporter]+rna[reporter]
Kf=k_forward * dna_reporter
k_forward=0.01
found_key=(mech=None, partid=None, name=kleak).
search_key=(mech=negativehill_transcription, partid=P_repressible_A, name=kleak).
2. rna[reporter] --> rna[reporter]+protein[reporter]
Kf=k_forward * rna_reporter
k_forward=0.25
found_key=(mech=simple_translation, partid=None, name=ktl).
search_key=(mech=simple_translation, partid=Strong, name=ktl).
3. rna[reporter] -->
Kf=k_forward * rna_reporter
k_forward=0.001
found_key=(mech=rna_degradation, partid=None, name=kdil).
search_key=(mech=rna_degradation, partid=rna_reporter, name=kdil).
]
/Users/murray/Library/CloudStorage/Dropbox/macosx/src/biocrnpyler/biocrnpyler/core/parameter.py:678: UserWarning: parameter file contains no unit column! Please add a column named ['unit', 'units'].
warn(
[4]:
#Titrate the repressor and plot the result
try:
import bioscrape
except ModuleNotFoundError:
print('please install the plotting libraries: pip install biocrnpyler[all]')
else:
import numpy as np
import pylab as plt
import pandas as pd
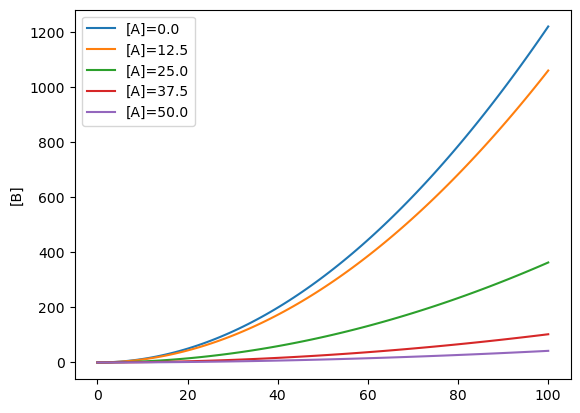
for r_c in np.linspace(0, 50, 5):
x0 = {repressible_assembly.dna:1, repressor:r_c}
timepoints = np.linspace(0, 100, 100)
R = CRN.simulate_with_bioscrape_via_sbml(timepoints, initial_condition_dict = x0)
plt.plot(R["time"], R[str(reporter)], label = f"[{str(S_A)}]={r_c}")
plt.ylabel("[B]")
plt.legend()
plt.show()
Example 3: A Simple Genetic Regulatory Network
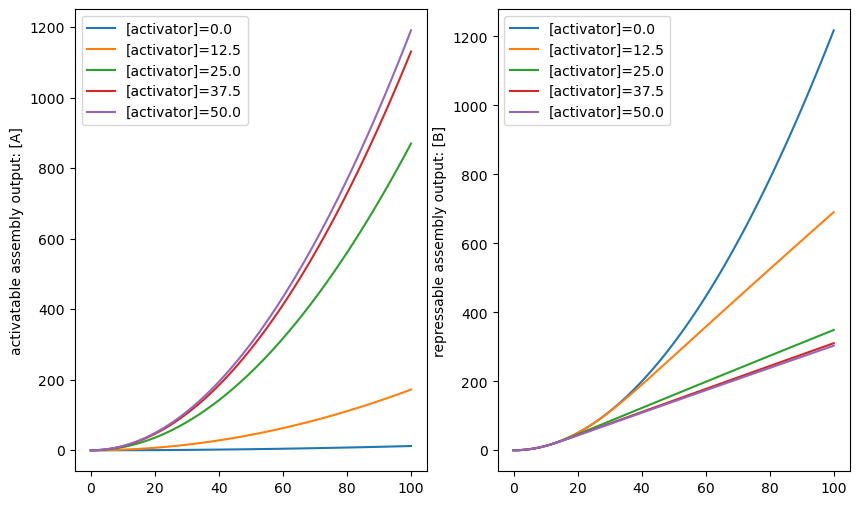
In this example, the activatable_assembly will produce a repressor that represses the repressable_assembly. Notice that activatable_assembly already procues the repressor of the RepressablePromoter…so this is easy!
[5]:
M = SimpleTxTlExtract(name="SimpleTxTl", parameter_file = "default_parameters.txt", components=[repressible_assembly, activatable_assembly])
CRN = M.compile_crn()
print(CRN.pretty_print(show_rates = True, show_keys = False))
#Titrate the activator, which in turn will automatically produce the repressor
try:
import bioscrape
except ModuleNotFoundError:
print('please install the plotting libraries: pip install biocrnpyler[all]')
else:
import numpy as np
import pylab as plt
import pandas as pd
plt.figure(figsize = (10, 6))
ax1, ax2 = plt.subplot(121), plt.subplot(122)#Create two subplots
for a_c in np.linspace(0, 50, 5):
x0 = {activatable_assembly.dna:1, repressible_assembly.dna:1, activator:a_c}
timepoints = np.linspace(0, 100, 100)
R = CRN.simulate_with_bioscrape_via_sbml(timepoints, initial_condition_dict = x0)
plt.sca(ax1)
plt.plot(R["time"], R[str(S_A)], label = "[activator]="+str(a_c))
plt.sca(ax2)
plt.plot(R["time"], R[str(reporter)], label = "[activator]="+str(a_c))
plt.sca(ax1)
plt.ylabel("activatable assembly output: [A]")
plt.legend()
plt.sca(ax2)
plt.ylabel("repressable assembly output: [B]")
plt.legend()
plt.show()
/Users/murray/Library/CloudStorage/Dropbox/macosx/src/biocrnpyler/biocrnpyler/core/parameter.py:678: UserWarning: parameter file contains no unit column! Please add a column named ['unit', 'units'].
warn(
Species(N = 7) = {
protein[reporter] (@ 0),
rna[reporter] (@ 0),
dna[reporter] (@ 0),
small_molecule[activator] (@ 0),
rna[activatable_assembly] (@ 0),
dna[activatable_assembly] (@ 0),
A (@ 0),
}
Reactions (8) = [
0. dna[reporter] --> dna[reporter]+rna[reporter]
Kf = k dna[reporter] / ( 1 + (A/K)^4 )
k=1.0
K=20
n=4
1. dna[reporter] --> dna[reporter]+rna[reporter]
Kf=k_forward * dna_reporter
k_forward=0.01
2. rna[reporter] --> rna[reporter]+protein[reporter]
Kf=k_forward * rna_reporter
k_forward=0.25
3. dna[activatable_assembly] --> dna[activatable_assembly]+rna[activatable_assembly]
Kf = k dna[activatable_assembly] small_molecule[activator]^n / ( K^n + small_molecule[activator]^n )
k=1.0
K=20
n=4
4. dna[activatable_assembly] --> dna[activatable_assembly]+rna[activatable_assembly]
Kf=k_forward * dna_activatable_assembly
k_forward=0.01
5. rna[activatable_assembly] --> rna[activatable_assembly]+A
Kf=k_forward * rna_activatable_assembly
k_forward=0.25
6. rna[reporter] -->
Kf=k_forward * rna_reporter
k_forward=0.001
7. rna[activatable_assembly] -->
Kf=k_forward * rna_activatable_assembly
k_forward=0.001
]
Example 4: RegulatedPromoter
In the below example, a CRN from RegulatedPromoter is generated. This Component models the detailed binding of regulators to the DNA and has seperate transcription rates for each regulator. It is suitable for complex models that include machinery. Regulators do not act Combinatorically.
[6]:
from biocrnpyler.core import ParameterKey
from biocrnpyler.components import RegulatedPromoter
from biocrnpyler.mixtures import TxTlExtract
#1 Regulated Promoter Needs lots of parameters!
component_parameters = {
#Promoter Activator Binding Parameters. Note the part_id = [promoter_name]_[regulator_name]
ParameterKey(mechanism = 'binding', part_id = 'regulated_promoter_A', name = 'kb'):100, #Promoter - Activator Binding
ParameterKey(mechanism = 'binding', part_id = "regulated_promoter_A", name = 'ku'):5.0, #Unbinding
ParameterKey(mechanism = 'binding', part_id = "regulated_promoter_A", name = 'cooperativity'):4.0, #Cooperativity
#Activated Promoter Transcription. Note the part_id = [promoter_name]_[regulator_name]
#These regulate RNAP binding to an activated promoter and transcription
ParameterKey(mechanism = 'transcription', part_id = 'regulated_promoter_A', name = 'kb'):100, #Promoter - Activator Binding
ParameterKey(mechanism = 'transcription', part_id = "regulated_promoter_A", name = 'ku'):1.0, #Unbinding
ParameterKey(mechanism = 'transcription', part_id = 'regulated_promoter_A', name = "ktx"): 1., #Transcription Rate
#Promoter Repressor Binding Parameters. Note the part_id = [promoter_name]_[regulator_name]
ParameterKey(mechanism = 'binding', part_id = 'regulated_promoter_R', name = 'kb'):100,
ParameterKey(mechanism = 'binding', part_id = "regulated_promoter_R", name = 'ku'):5.0,
ParameterKey(mechanism = 'binding', part_id = "regulated_promoter_R", name = 'cooperativity'):4.0,
#Repressed Promoter Transcription. Note the part_id = [promoter_name]_[regulator_name]
#These regulate RNAP binding to a repressed promoter and transcription
ParameterKey(mechanism = 'transcription', part_id = 'regulated_promoter_R', name = 'kb'):1,
ParameterKey(mechanism = 'transcription', part_id = "regulated_promoter_R", name = 'ku'):100.0,
ParameterKey(mechanism = 'transcription', part_id = 'regulated_promoter_R', name = "ktx"): 1.0, #Transcription Rate
#Leak Parameters for transcription
#These regulate expression of an unbound promoter
ParameterKey(mechanism = 'transcription', part_id = 'regulated_promoter_leak', name = "kb"): 2.,
ParameterKey(mechanism = 'transcription', part_id = 'regulated_promoter_leak', name = "ku"): 100,
ParameterKey(mechanism = 'transcription', part_id = 'regulated_promoter_leak', name = "ktx"): 1.0, #Transcription Rate
}
repressor = Species("R", material_type = "protein")
activator = Species("A", material_type = "protein")
reporter = Species("reporter", material_type = "protein")
#Create a RegulatedPromoter Object named "P_reg" with regulators "activator" and "repressor"
#By Loading custom parameters into the promoter, we override the default parameters of the Mixture
P_reg = RegulatedPromoter("regulated_promoter", regulators=[activator, repressor], leak=True, parameters = component_parameters)
#Create a DNA assembly "reporter" with P_reg for its promoter
reg_reporter = DNAassembly(name="reporter", promoter=P_reg, rbs="Strong", protein = reporter)
#Use a simple TxTl model with dilution
#M = SimpleTxTlDilutionMixture(name="e coli", parameter_file = "default_parameters.txt", components=[reg_reporter])
M = TxTlExtract(name="e coli extract", parameter_file = "default_parameters.txt", components=[reg_reporter])
CRN = M.compile_crn()
print(CRN.pretty_print(show_rates = True, show_keys = False))
/Users/murray/Library/CloudStorage/Dropbox/macosx/src/biocrnpyler/biocrnpyler/core/parameter.py:678: UserWarning: parameter file contains no unit column! Please add a column named ['unit', 'units'].
warn(
Species(N = 16) = {
protein[Ribo] (@ 24.0),
protein[RNAase] (@ 6.0),
protein[RNAP] (@ 3.0),
protein[reporter] (@ 0),
rna[reporter] (@ 0),
dna[reporter] (@ 0),
complex[protein[Ribo]:rna[reporter]] (@ 0),
complex[protein[RNAase]:rna[reporter]] (@ 0),
complex[dna[reporter]:2x_protein[R]] (@ 0),
complex[dna[reporter]:protein[RNAP]] (@ 0),
complex[dna[reporter]:2x_protein[A]] (@ 0),
complex[complex[protein[Ribo]:rna[reporter]]:protein[RNAase]] (@ 0),
complex[complex[dna[reporter]:2x_protein[R]]:protein[RNAP]] (@ 0),
complex[complex[dna[reporter]:2x_protein[A]]:protein[RNAP]] (@ 0),
protein[R] (@ 0),
protein[A] (@ 0),
}
Reactions (14) = [
0. dna[reporter]+protein[RNAP] <--> complex[dna[reporter]:protein[RNAP]]
Kf=k_forward * dna_reporter * protein_RNAP
Kr=k_reverse * complex_dna_reporter_protein_RNAP_
k_forward=2.0
k_reverse=100
1. complex[dna[reporter]:protein[RNAP]] --> dna[reporter]+rna[reporter]+protein[RNAP]
Kf=k_forward * complex_dna_reporter_protein_RNAP_
k_forward=1.0
2. 2protein[A]+dna[reporter] <--> complex[dna[reporter]:2x_protein[A]]
Kf=k_forward * protein_A^2 * dna_reporter
Kr=k_reverse * complex_dna_reporter_protein_A_2x_
k_forward=100.0
k_reverse=10.0
3. complex[dna[reporter]:2x_protein[A]]+protein[RNAP] <--> complex[complex[dna[reporter]:2x_protein[A]]:protein[RNAP]]
Kf=k_forward * complex_dna_reporter_protein_A_2x_ * protein_RNAP
Kr=k_reverse * complex_complex_dna_reporter_protein_A_2x__protein_RNAP_
k_forward=100
k_reverse=1.0
4. complex[complex[dna[reporter]:2x_protein[A]]:protein[RNAP]] --> complex[dna[reporter]:2x_protein[A]]+rna[reporter]+protein[RNAP]
Kf=k_forward * complex_complex_dna_reporter_protein_A_2x__protein_RNAP_
k_forward=1.0
5. 2protein[R]+dna[reporter] <--> complex[dna[reporter]:2x_protein[R]]
Kf=k_forward * protein_R^2 * dna_reporter
Kr=k_reverse * complex_dna_reporter_protein_R_2x_
k_forward=100.0
k_reverse=10.0
6. complex[dna[reporter]:2x_protein[R]]+protein[RNAP] <--> complex[complex[dna[reporter]:2x_protein[R]]:protein[RNAP]]
Kf=k_forward * complex_dna_reporter_protein_R_2x_ * protein_RNAP
Kr=k_reverse * complex_complex_dna_reporter_protein_R_2x__protein_RNAP_
k_forward=1
k_reverse=100.0
7. complex[complex[dna[reporter]:2x_protein[R]]:protein[RNAP]] --> complex[dna[reporter]:2x_protein[R]]+rna[reporter]+protein[RNAP]
Kf=k_forward * complex_complex_dna_reporter_protein_R_2x__protein_RNAP_
k_forward=1.0
8. rna[reporter]+protein[Ribo] <--> complex[protein[Ribo]:rna[reporter]]
Kf=k_forward * rna_reporter * protein_Ribo
Kr=k_reverse * complex_protein_Ribo_rna_reporter_
k_forward=100.0
k_reverse=10.0
9. complex[protein[Ribo]:rna[reporter]] --> rna[reporter]+protein[reporter]+protein[Ribo]
Kf=k_forward * complex_protein_Ribo_rna_reporter_
k_forward=0.05
10. complex[protein[Ribo]:rna[reporter]]+protein[RNAase] <--> complex[complex[protein[Ribo]:rna[reporter]]:protein[RNAase]]
Kf=k_forward * complex_protein_Ribo_rna_reporter_ * protein_RNAase
Kr=k_reverse * complex_complex_protein_Ribo_rna_reporter__protein_RNAase_
k_forward=100.0
k_reverse=10.0
11. complex[complex[protein[Ribo]:rna[reporter]]:protein[RNAase]] --> protein[Ribo]+protein[RNAase]
Kf=k_forward * complex_complex_protein_Ribo_rna_reporter__protein_RNAase_
k_forward=0.001
12. rna[reporter]+protein[RNAase] <--> complex[protein[RNAase]:rna[reporter]]
Kf=k_forward * rna_reporter * protein_RNAase
Kr=k_reverse * complex_protein_RNAase_rna_reporter_
k_forward=100.0
k_reverse=10.0
13. complex[protein[RNAase]:rna[reporter]] --> protein[RNAase]
Kf=k_forward * complex_protein_RNAase_rna_reporter_
k_forward=0.001
]
[7]:
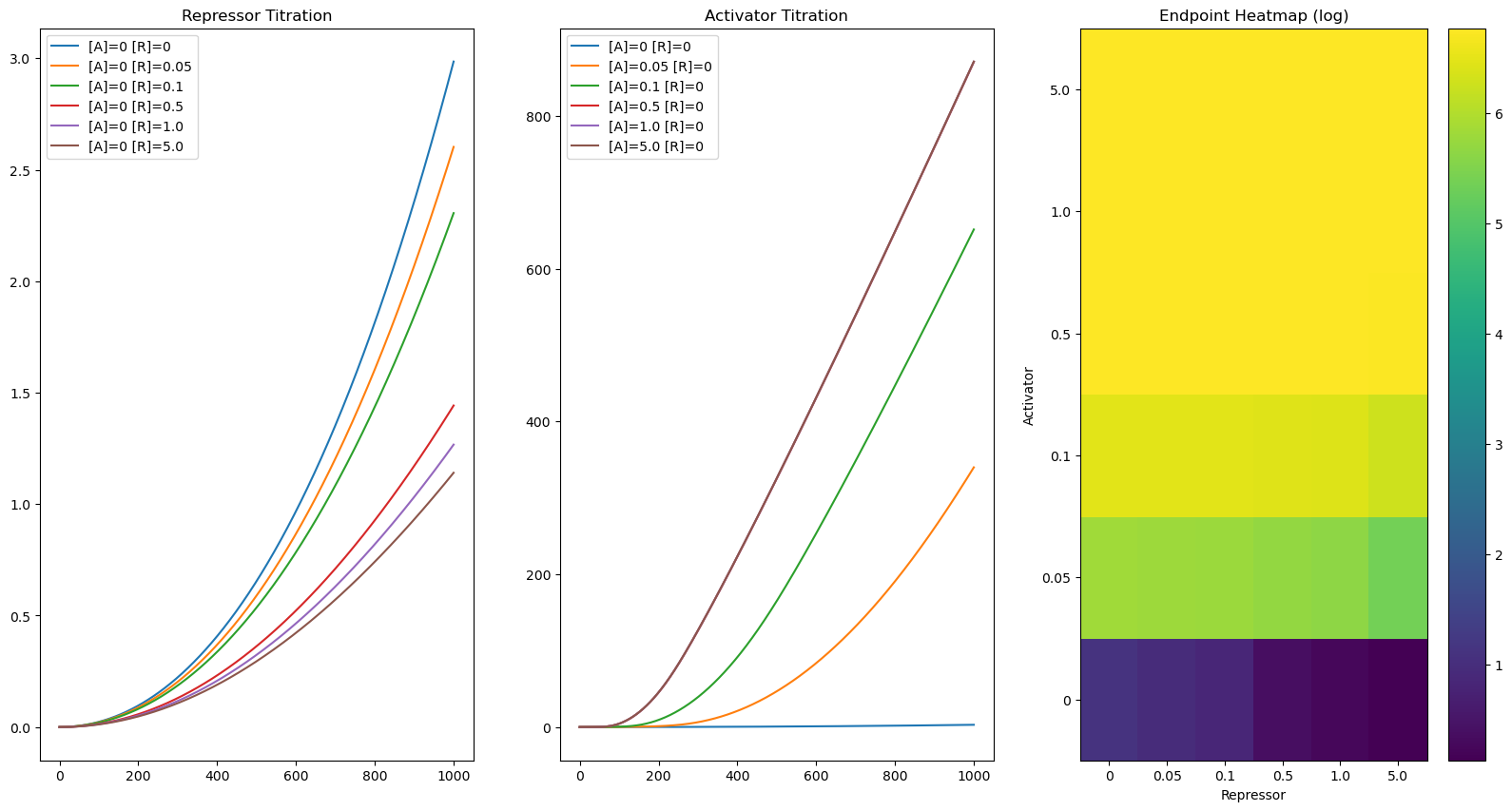
#Lets titrate Repressor and Activator - notice the bahvior is not combinatorial
try:
import bioscrape
except ModuleNotFoundError:
print('please install the plotting libraries: pip install biocrnpyler[all]')
else:
import numpy as np
import pylab as plt
import pandas as pd
plt.figure(figsize = (20, 10))
ax1, ax2, ax3 = plt.subplot(131), plt.subplot(132), plt.subplot(133)
titration_list = [0, .05, .1, .5, 1.0, 5.]
N = len(titration_list)
HM = np.zeros((N, N))
for a_ind, a_c in enumerate(titration_list):
for r_ind, r_c in enumerate(titration_list):
x0 = {reg_reporter.dna:.1, repressor:r_c, activator:a_c}
timepoints = np.linspace(0, 1000, 1000)
R = CRN.simulate_with_bioscrape_via_sbml(timepoints, initial_condition_dict = x0)
if a_ind == 0:
plt.sca(ax1)
plt.plot(R["time"], R[str(reporter)], label = "[A]="+str(a_c) +" [R]="+str(r_c))
if r_ind == 0:
plt.sca(ax2)
plt.plot(R["time"], R[str(reporter)], label = "[A]="+str(a_c) +" [R]="+str(r_c))
HM[a_ind, r_ind] = R[str(reporter)][len(timepoints)-1]
plt.sca(ax1)
plt.title("Repressor Titration")
plt.legend()
plt.sca(ax2)
plt.title("Activator Titration")
plt.legend()
plt.sca(ax3)
plt.title("Endpoint Heatmap (log)")
cb = plt.pcolor(np.log(HM))
plt.colorbar(cb)
plt.xlabel("Repressor")
plt.ylabel("Activator")
plt.xticks(np.arange(.5, N+.5, 1), [str(i) for i in titration_list])
plt.yticks(np.arange(.5, N+.5, 1), [str(i) for i in titration_list])
plt.show()
Example 5: Induction Model of a Ligand which Activates a Transcription Factor
In many biological circuits, small molecules (ligands) can bind to a transcription factor modulating its functionality.
In BioCRNpyler, we will model this by creating a ChemicalComplex Component which consists of a Transcription Factor and a ligand. This the ComplexSpecies formed by by binding the transcription will also work as the regulator (activator or repressor) of a regulated promoter. In this example, we will use RepressablePromoter.
In the activating case, the bound form of the ChemicalComplex will induce gene expression.
[8]:
from biocrnpyler.components import ChemicalComplex
from biocrnpyler.mixtures import ExpressionDilutionMixture
inactive_repressor = Species("A", material_type = "protein")
ligand = Species("L", material_type = "ligand")
#Create a ChemicalComplex to model ligand-inactive_repressor bindning
activatable_repressor = ChemicalComplex([inactive_repressor, ligand])
#Other Promoters could also be used
P_repressible = RepressiblePromoter("P_repressible", repressor = activatable_repressor.get_species(), leak = True, parameters = hill_parameters)
#Create a DNA assembly "reporter" with P_activatable for its promoter
repressible_assembly = DNAassembly(name="reporter", promoter=P_repressible, rbs="Strong", protein = "reporter")
M = ExpressionDilutionMixture(name="ExpressionDilutionMixture", parameter_file = "default_parameters.txt", components=[repressible_assembly, activatable_repressor])
CRN = M.compile_crn();print(CRN.pretty_print(show_rates = True, show_keys = False))
Species(N = 5) = {
protein[reporter] (@ 0),
dna[reporter] (@ 0),
complex[ligand[L]:protein[A]] (@ 0),
ligand[L] (@ 0),
protein[A] (@ 0),
}
Reactions (7) = [
0. dna[reporter] --> dna[reporter]+protein[reporter]
Kf = k dna[reporter] / ( 1 + (complex[ligand[L]:protein[A]]/K)^4 )
k=1.0
K=20
n=4
1. dna[reporter] --> dna[reporter]+protein[reporter]
Kf=k_forward * dna_reporter
k_forward=0.01
2. ligand[L]+protein[A] <--> complex[ligand[L]:protein[A]]
Kf=k_forward * ligand_L * protein_A
Kr=k_reverse * complex_ligand_L_protein_A_
k_forward=100.0
k_reverse=10.0
3. protein[reporter] -->
Kf=k_forward * protein_reporter
k_forward=0.001
4. ligand[L] -->
Kf=k_forward * ligand_L
k_forward=0.001
5. protein[A] -->
Kf=k_forward * protein_A
k_forward=0.001
6. complex[ligand[L]:protein[A]] -->
Kf=k_forward * complex_ligand_L_protein_A_
k_forward=0.001
]
/Users/murray/Library/CloudStorage/Dropbox/macosx/src/biocrnpyler/biocrnpyler/core/parameter.py:678: UserWarning: parameter file contains no unit column! Please add a column named ['unit', 'units'].
warn(
[9]:
#Lets titrate ligand and repressor
try:
import bioscrape
except ModuleNotFoundError:
print('please install the plotting libraries: pip install biocrnpyler[all]')
else:
import numpy as np
import pylab as plt
import pandas as pd
plt.figure(figsize = (8, 6))
N = 11 #Number of titrations
max_titration = 100
HM = np.zeros((N, N))
for r_ind, R_c in enumerate(np.linspace(0, max_titration, N)):
for l_ind, L_c in enumerate(np.linspace(0, max_titration, N)):
x0 = {repressible_assembly.dna:1, inactive_repressor:R_c, ligand:L_c}
timepoints = np.linspace(0, 1000, 1000)
R = CRN.simulate_with_bioscrape_via_sbml(timepoints, initial_condition_dict = x0)
HM[r_ind, l_ind] = R["protein_reporter"][len(timepoints)-1]
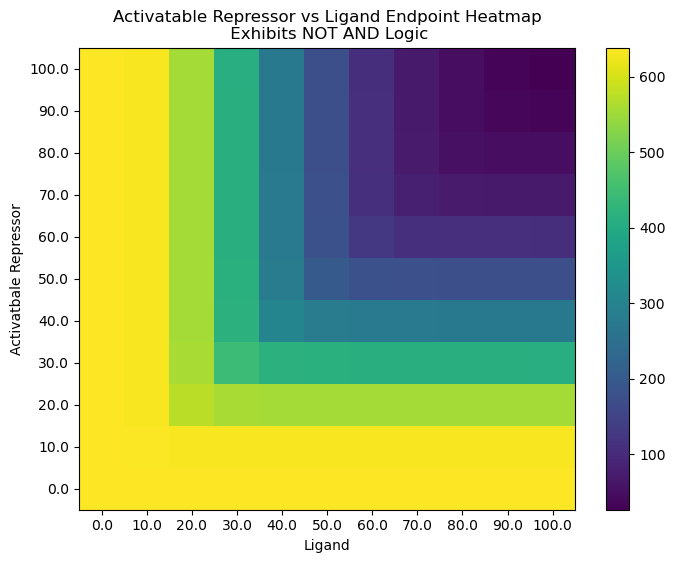
plt.title("Activatable Repressor vs Ligand Endpoint Heatmap\n Exhibits NOT AND Logic")
cb = plt.pcolor(HM)
plt.colorbar(cb)
plt.xlabel("Ligand")
plt.ylabel("Activatbale Repressor")
plt.xticks(np.arange(.5, N+.5, 1), [str(i) for i in np.linspace(0, max_titration, N)])
plt.yticks(np.arange(.5, N+.5, 1), [str(i) for i in np.linspace(0, max_titration, N)])
plt.show()
Example 6: Induction Models of a Ligand which Deactivates a Transcription Factor
In the inactivating case, the unbound transcription factor will activate the gene and the bound form will not.
[10]:
repressor = Species("A", material_type = "protein")
ligand = Species("L", material_type = "ligand")
#Create a ChemicalComplex to model ligand-inactive_repressor bindning
inactive_repressor = ChemicalComplex([repressor, ligand])
#Other Promoters could also be Used
P_repressible = RepressiblePromoter("P_repressible", repressor = repressor, leak = True, parameters = hill_parameters)
#Create a DNA assembly "reporter" with P_activatable for its promoter
repressible_assembly = DNAassembly(name="reporter", promoter=P_repressible, rbs="Strong", protein = "reporter")
M = ExpressionDilutionMixture(name="ExpressionDilutionMixture", parameter_file = "default_parameters.txt", components=[repressible_assembly, activatable_repressor])
CRN = M.compile_crn();print(CRN.pretty_print(show_rates = True, show_keys = False))
Species(N = 5) = {
protein[reporter] (@ 0),
dna[reporter] (@ 0),
complex[ligand[L]:protein[A]] (@ 0),
ligand[L] (@ 0),
protein[A] (@ 0),
}
Reactions (7) = [
0. dna[reporter] --> dna[reporter]+protein[reporter]
Kf = k dna[reporter] / ( 1 + (protein[A]/K)^4 )
k=1.0
K=20
n=4
1. dna[reporter] --> dna[reporter]+protein[reporter]
Kf=k_forward * dna_reporter
k_forward=0.01
2. ligand[L]+protein[A] <--> complex[ligand[L]:protein[A]]
Kf=k_forward * ligand_L * protein_A
Kr=k_reverse * complex_ligand_L_protein_A_
k_forward=100.0
k_reverse=10.0
3. protein[reporter] -->
Kf=k_forward * protein_reporter
k_forward=0.001
4. ligand[L] -->
Kf=k_forward * ligand_L
k_forward=0.001
5. protein[A] -->
Kf=k_forward * protein_A
k_forward=0.001
6. complex[ligand[L]:protein[A]] -->
Kf=k_forward * complex_ligand_L_protein_A_
k_forward=0.001
]
/Users/murray/Library/CloudStorage/Dropbox/macosx/src/biocrnpyler/biocrnpyler/core/parameter.py:678: UserWarning: parameter file contains no unit column! Please add a column named ['unit', 'units'].
warn(
[11]:
#Titration of ligand and repressor
try:
import bioscrape
except ModuleNotFoundError:
print('please install the plotting libraries: pip install biocrnpyler[all]')
else:
import numpy as np
import pylab as plt
import pandas as pd
plt.figure(figsize = (8, 6))
N = 11 #Number of titrations
max_titration = 100
HM = np.zeros((N, N))
for r_ind, R_c in enumerate(np.linspace(0, max_titration, N)):
for l_ind, L_c in enumerate(np.linspace(0, max_titration, N)):
x0 = {repressible_assembly.dna:1, repressor:R_c, ligand:L_c}
timepoints = np.linspace(0, 1000, 1000)
R = CRN.simulate_with_bioscrape_via_sbml(timepoints, initial_condition_dict = x0)
HM[r_ind, l_ind] = R["protein_reporter"][len(timepoints)-1]
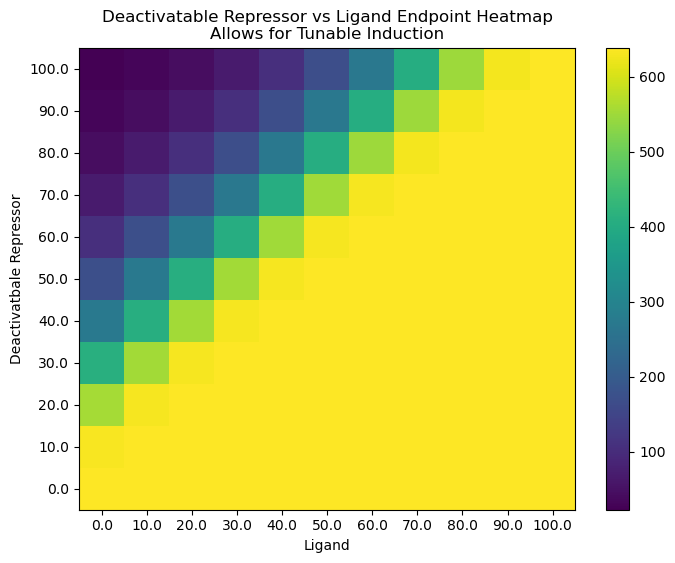
plt.title("Deactivatable Repressor vs Ligand Endpoint Heatmap\nAllows for Tunable Induction")
cb = plt.pcolor(HM)
plt.colorbar(cb)
plt.xlabel("Ligand")
plt.ylabel("Deactivatbale Repressor")
plt.xticks(np.arange(.5, N+.5, 1), [str(i) for i in np.linspace(0, max_titration, N)])
plt.yticks(np.arange(.5, N+.5, 1), [str(i) for i in np.linspace(0, max_titration, N)])
plt.show()
Example 7: Modeling AND, OR, and XOR Promoters with Combinatorial Promoter
CombinatorialPromoter is a Component designed to model arbitrary combinatorial logic on a promoter. For example, a promoter with 2 transcription factor binding sites can have 4 differents states:
Nothing bound
just factor 1 bound
just factor 2 bound
factors 1 and 2 bound
In general, a promoter with \(N\) binding sites has up to \(2^N\) possible states. Combinatorial promoter enumerates all these states and allows for the modeller to decide which are capable of transcription and which are not. For more details on this class, see the CombinatorialPromoter example ipython notebook in the BioCRNpyler examples folder.
Below, we will use a Combinatorial Promoter to Produce OR, AND, and XOR logic with two species, \(A\) and \(B\) by passing in lists of the transcribable combinations of regulators to the tx_capable_list keyword.
[12]:
from biocrnpyler.components import CombinatorialPromoter
from biocrnpyler.mixtures import ExpressionExtract
#AND Logic
A = Species("A") ;B = Species("B") #Inducers
#Create the Combinatorial Promoter
Prom_AND = CombinatorialPromoter("combinatorial_promoter",[A,B], tx_capable_list = [[A,B]], leak = True) #the Combination A and B can be transcribed
AND_assembly = DNAassembly("AND",promoter=Prom_AND,rbs="medium",protein="GFP")
#Use an Expression Mixture to focus on Logic, not Transcription & Translation
M = ExpressionExtract(name="expression", parameter_file = "default_parameters.txt", components=[AND_assembly])
CRN = M.compile_crn(); print(CRN.pretty_print(show_rates = True, show_keys = False))
#Lets titrate A and B
try:
import bioscrape
except ModuleNotFoundError:
print('please install the plotting libraries: pip install biocrnpyler[all]')
else:
import numpy as np
import pylab as plt
import pandas as pd
plt.figure(figsize = (8, 6))
N = 11 #Number of titrations
max_titration = 10
HM = np.zeros((N, N))
for a_ind, A_c in enumerate(np.linspace(0, max_titration, N)):
for b_ind, B_c in enumerate(np.linspace(0, max_titration, N)):
x0 = {AND_assembly.dna:1, A:A_c, B:B_c}
timepoints = np.linspace(0, 1000, 1000)
R = CRN.simulate_with_bioscrape_via_sbml(timepoints, initial_condition_dict = x0)
HM[a_ind, b_ind] = R["protein_GFP"][len(timepoints)-1]
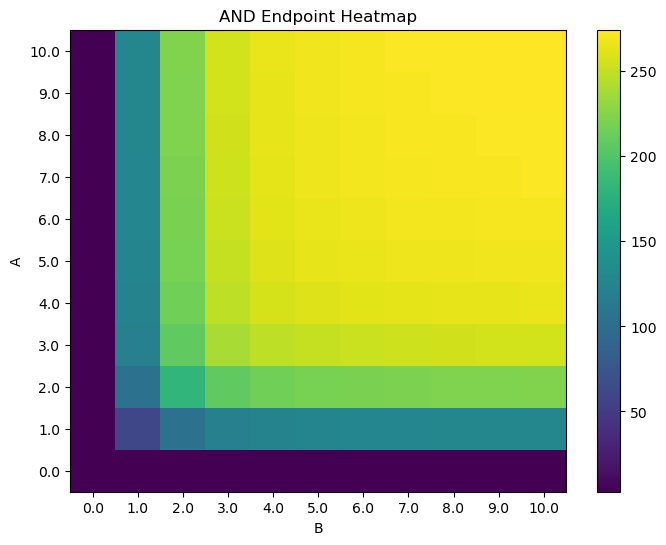
plt.title("AND Endpoint Heatmap")
cb = plt.pcolor(HM)
plt.colorbar(cb)
plt.xlabel("B")
plt.ylabel("A")
plt.xticks(np.arange(.5, N+.5, 1), [str(i) for i in np.linspace(0, max_titration, N)])
plt.yticks(np.arange(.5, N+.5, 1), [str(i) for i in np.linspace(0, max_titration, N)])
plt.show()
/Users/murray/Library/CloudStorage/Dropbox/macosx/src/biocrnpyler/biocrnpyler/core/parameter.py:678: UserWarning: parameter file contains no unit column! Please add a column named ['unit', 'units'].
warn(
Species(N = 7) = {
protein[GFP] (@ 0),
complex[2x_B:dna[AND]] (@ 0),
B (@ 0),
complex[2x_A:dna[AND]] (@ 0),
complex[2x_A:2x_B:dna[AND]] (@ 0),
dna[AND] (@ 0),
A (@ 0),
}
Reactions (8) = [
0. dna[AND] --> dna[AND]+protein[GFP]
Kf=k_forward * dna_AND
k_forward=0.0028125
1. 2A+dna[AND] <--> complex[2x_A:dna[AND]]
Kf=k_forward * A^2 * dna_AND
Kr=k_reverse * complex_A_2x_dna_AND_
k_forward=100.0
k_reverse=10.0
2. 2B+dna[AND] <--> complex[2x_B:dna[AND]]
Kf=k_forward * B^2 * dna_AND
Kr=k_reverse * complex_B_2x_dna_AND_
k_forward=100.0
k_reverse=10.0
3. 2A+complex[2x_B:dna[AND]] <--> complex[2x_A:2x_B:dna[AND]]
Kf=k_forward * A^2 * complex_B_2x_dna_AND_
Kr=k_reverse * complex_A_2x_B_2x_dna_AND_
k_forward=100.0
k_reverse=10.0
4. 2B+complex[2x_A:dna[AND]] <--> complex[2x_A:2x_B:dna[AND]]
Kf=k_forward * B^2 * complex_A_2x_dna_AND_
Kr=k_reverse * complex_A_2x_B_2x_dna_AND_
k_forward=100.0
k_reverse=10.0
5. complex[2x_A:2x_B:dna[AND]] --> complex[2x_A:2x_B:dna[AND]]+protein[GFP]
Kf=k_forward * complex_A_2x_B_2x_dna_AND_
k_forward=0.28125
6. complex[2x_A:dna[AND]] --> complex[2x_A:dna[AND]]+protein[GFP]
Kf=k_forward * complex_A_2x_dna_AND_
k_forward=0.0028125
7. complex[2x_B:dna[AND]] --> complex[2x_B:dna[AND]]+protein[GFP]
Kf=k_forward * complex_B_2x_dna_AND_
k_forward=0.0028125
]
[13]:
#Create OR Logic
Prom_OR = CombinatorialPromoter("combinatorial_promoter",[A,B], leak=False,
tx_capable_list = [[A,B], [A], [B]]) #the Combinations A and B or just A or just B be transcribed
ORassembly = DNAassembly("OR",promoter=Prom_OR,rbs="medium",protein="GFP")
print(ORassembly)
#Use an Expression Mixture to focus on Logic, not Transcription & Translation
M = ExpressionExtract(name="expression", parameter_file = "default_parameters.txt", components=[ORassembly])
CRN = M.compile_crn()
print(CRN.pretty_print(show_rates = True, show_keys = False))
#Lets titrate A and B
try:
import bioscrape
except ModuleNotFoundError:
print('please install the plotting libraries: pip install biocrnpyler[all]')
else:
import numpy as np
import pylab as plt
import pandas as pd
plt.figure(figsize = (6, 6))
N = 11 #Number of titrations
max_titration = 10
HM = np.zeros((N, N))
for a_ind, A_c in enumerate(np.linspace(0, max_titration, N)):
for b_ind, B_c in enumerate(np.linspace(0, max_titration, N)):
x0 = {ORassembly.dna:1, A:A_c, B:B_c}
timepoints = np.linspace(0, 1000, 1000)
R = CRN.simulate_with_bioscrape_via_sbml(timepoints, initial_condition_dict = x0)
HM[a_ind, b_ind] = R["protein_GFP"][len(timepoints)-1]
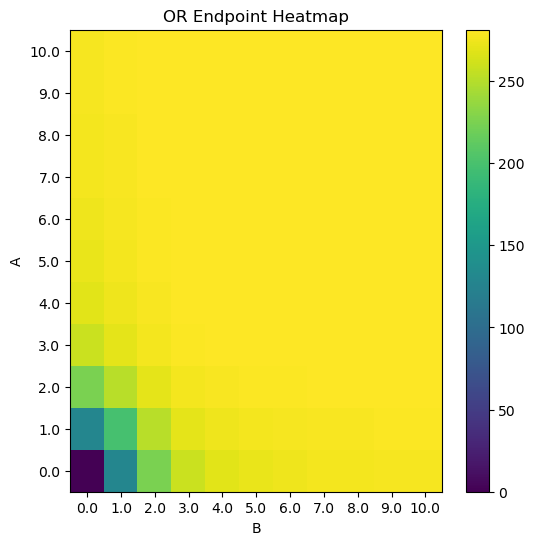
plt.title("OR Endpoint Heatmap")
cb = plt.pcolor(HM)
plt.colorbar(cb)
plt.xlabel("B")
plt.ylabel("A")
plt.xticks(np.arange(.5, N+.5, 1), [str(i) for i in np.linspace(0, max_titration, N)])
plt.yticks(np.arange(.5, N+.5, 1), [str(i) for i in np.linspace(0, max_titration, N)])
plt.show()
DNAassembly: OR
Species(N = 7) = {
dna[OR] (@ 0),
protein[GFP] (@ 0),
complex[2x_B:dna[OR]] (@ 0),
B (@ 0),
complex[2x_A:dna[OR]] (@ 0),
complex[2x_A:2x_B:dna[OR]] (@ 0),
A (@ 0),
}
Reactions (7) = [
0. 2A+dna[OR] <--> complex[2x_A:dna[OR]]
Kf=k_forward * A^2 * dna_OR
Kr=k_reverse * complex_A_2x_dna_OR_
k_forward=100.0
k_reverse=10.0
1. 2B+dna[OR] <--> complex[2x_B:dna[OR]]
Kf=k_forward * B^2 * dna_OR
Kr=k_reverse * complex_B_2x_dna_OR_
k_forward=100.0
k_reverse=10.0
2. 2A+complex[2x_B:dna[OR]] <--> complex[2x_A:2x_B:dna[OR]]
Kf=k_forward * A^2 * complex_B_2x_dna_OR_
Kr=k_reverse * complex_A_2x_B_2x_dna_OR_
k_forward=100.0
k_reverse=10.0
3. 2B+complex[2x_A:dna[OR]] <--> complex[2x_A:2x_B:dna[OR]]
Kf=k_forward * B^2 * complex_A_2x_dna_OR_
Kr=k_reverse * complex_A_2x_B_2x_dna_OR_
k_forward=100.0
k_reverse=10.0
4. complex[2x_A:dna[OR]] --> complex[2x_A:dna[OR]]+protein[GFP]
Kf=k_forward * complex_A_2x_dna_OR_
k_forward=0.28125
5. complex[2x_B:dna[OR]] --> complex[2x_B:dna[OR]]+protein[GFP]
Kf=k_forward * complex_B_2x_dna_OR_
k_forward=0.28125
6. complex[2x_A:2x_B:dna[OR]] --> complex[2x_A:2x_B:dna[OR]]+protein[GFP]
Kf=k_forward * complex_A_2x_B_2x_dna_OR_
k_forward=0.28125
]
/Users/murray/Library/CloudStorage/Dropbox/macosx/src/biocrnpyler/biocrnpyler/core/parameter.py:678: UserWarning: parameter file contains no unit column! Please add a column named ['unit', 'units'].
warn(
[14]:
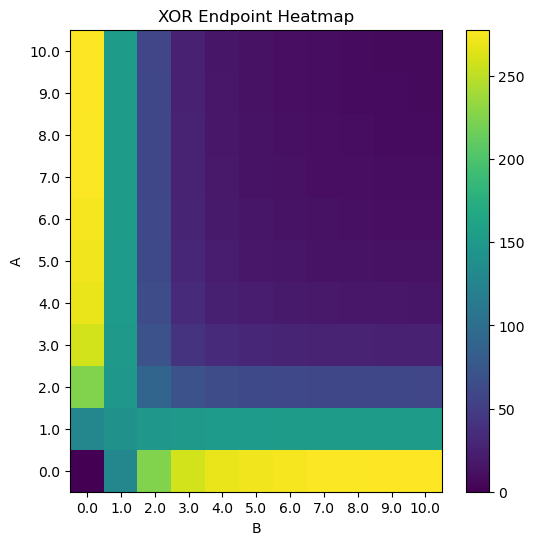
#Create XOR Logic
Prom_XOR = CombinatorialPromoter("combinatorial_promoter",[A,B], leak=False,
tx_capable_list = [[A], [B]]) #the Combinations just A or just B can be transcribed
XORassembly = DNAassembly("XOR",promoter=Prom_XOR,rbs="medium",protein="GFP")
#Use an Expression Mixture to focus on Logic, not Transcription & Translation
M = ExpressionExtract(name="expression", parameter_file = "default_parameters.txt", components=[XORassembly])
CRN = M.compile_crn()
print(CRN.pretty_print(show_rates = True, show_keys = False))
#Lets titrate A and B
try:
import bioscrape
except ModuleNotFoundError:
print('please install the plotting libraries: pip install biocrnpyler[all]')
else:
import numpy as np
import pylab as plt
import pandas as pd
plt.figure(figsize = (6, 6))
N = 11 #Number of titrations
max_titration = 10
HM = np.zeros((N, N))
for a_ind, A_c in enumerate(np.linspace(0, max_titration, N)):
for b_ind, B_c in enumerate(np.linspace(0, max_titration, N)):
x0 = {XORassembly.dna:1, A:A_c, B:B_c}
timepoints = np.linspace(0, 1000, 1000)
R = CRN.simulate_with_bioscrape_via_sbml(timepoints, initial_condition_dict = x0)
HM[a_ind, b_ind] = R["protein_GFP"][len(timepoints)-1]
plt.title("XOR Endpoint Heatmap")
cb = plt.pcolor(HM)
plt.colorbar(cb)
plt.xlabel("B")
plt.ylabel("A")
plt.xticks(np.arange(.5, N+.5, 1), [str(i) for i in np.linspace(0, max_titration, N)])
plt.yticks(np.arange(.5, N+.5, 1), [str(i) for i in np.linspace(0, max_titration, N)])
plt.show()
/Users/murray/Library/CloudStorage/Dropbox/macosx/src/biocrnpyler/biocrnpyler/core/parameter.py:678: UserWarning: parameter file contains no unit column! Please add a column named ['unit', 'units'].
warn(
Species(N = 7) = {
dna[XOR] (@ 0),
protein[GFP] (@ 0),
complex[2x_B:dna[XOR]] (@ 0),
B (@ 0),
complex[2x_A:dna[XOR]] (@ 0),
complex[2x_A:2x_B:dna[XOR]] (@ 0),
A (@ 0),
}
Reactions (6) = [
0. 2A+dna[XOR] <--> complex[2x_A:dna[XOR]]
Kf=k_forward * A^2 * dna_XOR
Kr=k_reverse * complex_A_2x_dna_XOR_
k_forward=100.0
k_reverse=10.0
1. 2B+dna[XOR] <--> complex[2x_B:dna[XOR]]
Kf=k_forward * B^2 * dna_XOR
Kr=k_reverse * complex_B_2x_dna_XOR_
k_forward=100.0
k_reverse=10.0
2. 2A+complex[2x_B:dna[XOR]] <--> complex[2x_A:2x_B:dna[XOR]]
Kf=k_forward * A^2 * complex_B_2x_dna_XOR_
Kr=k_reverse * complex_A_2x_B_2x_dna_XOR_
k_forward=100.0
k_reverse=10.0
3. 2B+complex[2x_A:dna[XOR]] <--> complex[2x_A:2x_B:dna[XOR]]
Kf=k_forward * B^2 * complex_A_2x_dna_XOR_
Kr=k_reverse * complex_A_2x_B_2x_dna_XOR_
k_forward=100.0
k_reverse=10.0
4. complex[2x_A:dna[XOR]] --> complex[2x_A:dna[XOR]]+protein[GFP]
Kf=k_forward * complex_A_2x_dna_XOR_
k_forward=0.28125
5. complex[2x_B:dna[XOR]] --> complex[2x_B:dna[XOR]]+protein[GFP]
Kf=k_forward * complex_B_2x_dna_XOR_
k_forward=0.28125
]
[15]:
# End